Summary Reports for MS/MS
At the completion of a search, a summary report is displayed that
provides an overview of the results. There is a choice of
report formats and all reports contain links to more detailed
views of the experimental and calculated data.
For searches of less than 300 MS/MS spectra, the default summary
report is the Peptide Summary. This provides a clear picture of the peptide matches,
grouped into protein hits using a simple parsimony algorithm.
If there are 300 or more spectra, the default summary
report is the Protein Family Summary. This groups the proteins into families
based on a novel hierarchical clustering algorithm and
presents these results one page at a time, initially with 10 families per page. This report is ideally
suited to very large and complex MS/MS searches, where it is not practical to display all the results on a single
HTML page. For additional information about the Protein Family Summary, see
Koskinen, V. R.,
Hierarchical Clustering of Shotgun Proteomics Data 2011 MCP 10 M110.003822
You can use the format controls to switch to a Select
Summary, which is similar to a Peptide Summary, but provides a more compact
view of the results. The Select Summary splits the peptide
matches assigned to protein hits into a separate report from
the unassigned peptide matches.
For searches of less than 1000 MS/MS spectra, you can also choose a Protein Summary,
but it is not recommended to do so
unless you are viewing the results of a combination search.
If the sample is a mixture, using one of the Protein Summary reports to view the
results can give a very misleading picture.
If you are submitting MS/MS searches to an in-house Mascot server, and display the
results as a Peptide Summary, you will also have the option
to create an Archive Report. This is simply an edited version of the Peptide Summary report,
that only includes the protein hits you have selected.
If there are no
peptide sequence matches whatsoever from a search of MS/MS data, only molecular weight matches,
then a Protein Summary
report will be displayed. This indicates that the search
has failed. Possibly the spectra are nothing but noise or possibly the search parameters
are incorrect in some way.
Sections of the report are described in the order in which they appear.
Use
this link to open an example report in a new browser window or tab.
(The Protein Family Summary makes extensive use of JavaScript and Scalable Vector
Graphics. For full functionality, use Mozilla Firefox 2.0.0.14 or later or the current
release of Google Chrome or Microsoft Internet
Explorer 6.0.2800.1106 or later with the Adobe SVG Viewer plug-in 3.0. Further
details on this page.)
Most of the information in a Family Summary is structured into sections that can be displayed or hidden.
Links that toggle between the two states are easily recognised by the adjacent
triangle. The triangle points to the right when the section is collapsed and points down when
the section is expanded.
At the top of the report are a few lines to identify the search uniquely:
search title, date, user name, etc. The database version is identified with either a release
number or an ISO datestamp.
A search can easily be repeated, so as to investigate the effect of changes in search
parameters. The choices for
repeating a search are set by radio buttons for All queries, Non-significant
(below identity threshold), and Unassigned.
A drop-down list contains available export formats. Select the required format and
choose Export. There is also a link to switch back to the earlier,
Select Summary report, in case you are using some third party
software application that only accepts the earlier report formats.
The search parameters can be displayed or hidden by clicking on the sub-heading.
Descriptions of individual search parameters can be found here.
Histograms illustrating the peptide and protein score distributions
can be displayed or hidden by clicking on the sub-heading.
The left hand histogram shows the peptide score distribution, divided into 16
bins. The heights of the bars show the number of matches in each bin.
Similarly, the right hand histogram shows the
50 highest protein scores. For a search of MS/MS data, protein scores are derived from ions scores as a
non-probabilistic basis for ranking protein hits. The protein score histogram has
little meaning for MS/MS results, but is retained for historical reasons.
A legend, explaining how font style and colour are used to convey information about individual
peptide matches can be displayed or hidden by clicking on the sub-heading.
Red indicates the top-ranking peptide match for the query. Bold indicates significant (score greater than
homology threshold). Note that this meaning of bold is different from that used in the Peptide Summary and
Select Summary. Italic font is used for a duplicate match (more than one query matches to the
same peptide sequence with the same modifications, and charge state).
These controls enable the report format to be modified. After making changes, press the
"Format As" button to reload the report using the new settings.
- Significance threshold The default significance threshold is p < 0.05. You can
change this to any value in the range 0.99 to 1E-18.
- Maximum number of families This value was initially chosen when the search was submitted.
Enter a positive integer if you wish to re-specify the number of protein families to report.
Of course, the total number of families actually found by the search may be less.
Entering the word AUTO or a value of 0 will display all of the families
that have at least one peptide match with a significant score.
- Ions score cut-off Values greater than 0 and less than 1 act as an expect value threshold,
and any peptide matches
with higher expect values are suppressed. Values of 1 or more act
as a score threshold, and any peptide matches with lower scores are suppressed. By setting this to (say) 0.05,
you cut out all of the non-significant peptide matches, making the report shorter and more readable.
- Dendrograms cut at Causes all dendrograms to be cut at the specified score.
The following controls are only displayed for certain types of search:
- UniGene index (Only displayed for a search of a nucleic acid database when UniGene index files have
been configured) Choose the UniGene index to be used to cluster the proteins
into gene based families.
- Error tolerant matches (Only displayed for an automatic error tolerant search)
A drop-down list can be used to switch between displaying both standard and error tolerant matches
(the default), displaying just the standard matches, and displaying just the error tolerant matches.
- Show Percolator scores (Only displayed for searches that satisfy the requirements for using
Percolator) Check to display scores and expect values adjusted by Percolator.
- Preferred taxonomy
If the search used a quantitation method specifying Multiplex or Reporter protocol,
there will be an additional block of quantitation related controls, as
described here. Checkboxes can be used to toggle the
display of protein and peptide ratios.
If the search included the auto-decoy option,
false discovery rate information is displayed at this location.
The body of the report consists of three tabs, one for protein families,
one for Report Builder, and one for
unassigned matches.
Proteins are grouped into families using a novel
hierarchical clustering algorithm.
If the family contains a single member, the accession string, protein score and description are listed. If
the family contains multiple members, the accessions, scores and descriptions are aligned with a dendrogram,
which illustrates the degree of similarity between members. To see complete information about
the proteins in the family, and the peptide matches assigned
to each family member, click on the family number link.
In the example result report,
family 3 has 3 members. To make the report more concise, set the
Ions score or expect cut-off to 0.05 and choose Filter. This will remove all the
low scoring matches. Now, expand family 3 by clicking on the family number link.
The dendrogram illustrates the degree of similarity between members of a protein family. The scale is
ions score, and HSP7C_MOUSE and HS71L_MOUSE join at a score of approximately 30. This represents the score
of the significant matches that would have to be discarded in order to make one protein a sub-set of the other.
These two proteins are much more similar
to one other than to GRP78_MOUSE, which has non-shared peptide matches with a total score of approximately 145.
Note that, where there are multiple matches to the same peptide sequence, (ignoring charge state and
modification state), it is the highest score for each sequence that is used.
Immediately under the dendrogram is a list of the proteins. In this example, because SwissProt has low
redundancy, each family member is a single protein. In other cases, a family member will represent multiple
same-set proteins. One of the proteins is chosen as the anchor protein, to be listed first,
and the other same-set proteins are collapsed under a same-set heading. There is nothing special about the
protein picked for the anchor position. You may have a preference for one according to taxonomy or description, but
all proteins in a same-set group are indistinguishable on the basis of the peptide match evidence.
Clicking on the accession string link will load a
Protein View report. For
each of the proteins, beside the protein score and mass, there are counts of the number of matches and
the number of distinct sequences. In each column, the first number is the total count while the number in
parentheses is the count for matches above the significance threshold. If you filtered the report to
include only matches above the threshold, as suggested, the two numbers will always be the same.
To see the peptides that distinguish HSP7C_MOUSE and HS71L_MOUSE,
clear the checkbox for GRP78_MOUSE and choose Redisplay. The table of peptide matches will be reduced to rows and columns for
just these two proteins, making it easier to compare the matches. A marker indicates that a particular peptide is found in a particular
protein. It can be seen that HS71L_MOUSE would be a sub-set of
HSP7C_MOUSE if it was not for one match, K.ATAGDTHLGGEDFDNR.L, which is present in HS71L_MOUSE and not in HSP7C_MOUSE.
It is the significant score for this match that separates the two proteins in the dendrogram by a distance of 32 (score of
55 - homology threshold score of 23).
If you look a little more closely, you will notice that K.STAGDTHLGGEDFDNR.M has a weak match in HSP7C_MOUSE. So, the
evidence for both proteins being present comes down to a single residue. If S221 in HSP7C_MOUSE was an A, or A223 in HS71L_MOUSE
was an S, then HS71L_MOUSE would be a sub-set protein. Interestingly, the stronger match is for the sequence found in the protein with fewer
matches. This could be chance or it could be that the analyte sequence was essentially HSP7C_MOUSE but with an A at this position.
You can "cut" the dendrogram using the controls underneath. The slider control may not work in all
browsers. If this is the case, you can type a numeric value into the threshold field. By using the slider or by entering a number,
set the threshold to 50 and choose Cut. HS71L_MOUSE will be dropped from the dendrogram and the peptide match table
because it is now a sub-set protein. If you compare the matches to HSP7C_MOUSE with those to GRP78_MOUSE, it becomes clear
that these are very different proteins. They are part of the same family because of two shared matches, LIGDAAK and IINEPTAAAIAYGLD,
but many highly significant matches would have to be discarded for either protein to become a sub-set of the other. In summary,
we can quickly deduce from the Family Summary that
there is abundant evidence that both GRP78_MOUSE and HSP7C_MOUSE were present in the sample. There is little evidence for HS71L_MOUSE.
It is more likely that the HSP7C_MOUSE contained a SNP or two relative to the database sequence.
The peptide match table contains the following columns:
- Query number, hyperlinked to Peptide View.
- Dupes is a count of the number of additional matches to the same peptide sequence with the same modifications
and charge. Click on the link to show these duplicate matches, which will have the same or lower score than the
one first listed
- Experimental m/z value
- Experimental m/z transformed to a relative molecular mass
- Relative molecular mass calculated from the matched peptide sequence
- Difference (error) between the experimental and calculated masses
- Number of missed cleavage sites
- Ions score - If there are duplicate matches to the same peptide, then
the lower scoring matches are shown in brackets.
- Expectation value for the peptide match. (The number of times we would expect to obtain an equal or
higher score, purely by chance. The lower this value, the more
significant the result).
- Rank of the peptide match, (1 to 10, where 1 is the best match). If there is a triangle
to the left of the rank, the row can be expanded to show the alternative
matches for this query.
- A letter U if the peptide sequence is unique to the protein family. (Since the family
is an exhaustive grouping of proteins related by significant peptide matches,
all significant matches will be labelled U. Non-significant matches may or may not
be unique.)
- A column is displayed for each family member selected by its checkbox immediately
above the table. A marker is shown if the peptide match is present in the protein.
Where a column represents more than one protein, and the peptide is found in some
of these proteins, but not all, the marker is grey. Otherwise, it is black.
- Sequence of the peptide in 1-letter code. The residues that
bracket the peptide sequence in the protein are also shown, delimited
by periods. If the peptide forms the protein terminus, then a dash
is shown instead.
- Any variable modifications used to obtain the match
If the peptide sequence is modified, each affected residue is underlined. Details of the
modification will be displayed if the mouse cursor is rested over the residue. If multiple
matches to a query have identical scores, i.e. the sequences
are different, but are identical in mass spectrometry terms, they are collapsed into a single
consensus peptide sequence for display. The residues that differ between the matches are
displayed in lower case. For example, if deamidation was a variable modification and
one protein contained the sequence FASFIDK and another protein in the same family contained
FASFINK (deamidation at N) then the table would display FASFIdK. Click on the rank
to expand the alternative matches for the query and the actual peptide sequences will be
displayed.
Controls at the top and bottom of the tab can be used to
move between pages and change the number of protein families per page.
There are also buttons to expand and collapse all of the information on the page.
At the top of the tab, there are text search controls.
You can search the report for proteins and peptides containing numerical or text
values.
Search for proteins by:
- Accession
- Description
- Family number
- Page number
Search for peptides by:
- Query number
- Observed m/z
- Mr(expt)
- Mr(calc)
- Sequence
- Fixed modification
- Variable modification
If the target is found, the first occurence will be highlighted and,
if necessary, the page will scroll and expand.
If the target is found in multiple locations in a
single family, all matches will be highlighted. Choose Next
or Previous to jump forward or backward to other instances of the target in other families.
If the target is not found, a button provides the option to try the search
in the Unassigned tab.
The Report Builder tab allows you to build a customised table of protein hits,
which is particularly useful if you need a minimal list of proteins
for a publication. You
can choose which columns to include and their order, filter out proteins that are of no
interest, such as one-hit wonders, and export the table in CSV format directly to Excel.
The table has one row for each of the top-level protein hits, sometimes called anchor
proteins. Since a family can contain several
family members, grouped because they have shared peptides, there will be as many
rows in the table as there are family members in the report.
If the search results contain same-set proteins, and a preferred
taxonomy has been selected, this will also apply to the selection of anchor
proteins for the table.
You can sort the table by clicking on a column header. The currently active sort order is
shown by an arrow in the relevant column; up means ascending, down means descending.
The table can be exported as CSV with one click, with sort order preserved.
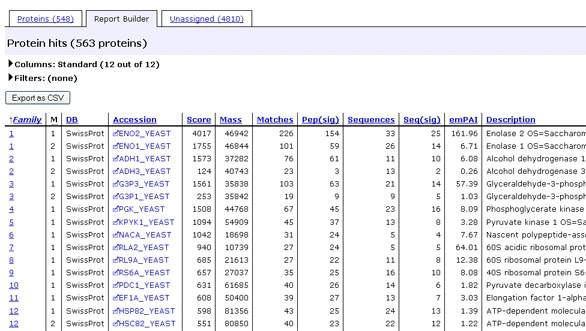
The columns are mostly self-explanatory, and tooltip help
is displayed when the mouse pointer rests over
a column header. Clicking on a family number hyperlink will jump to the
family displayed in the proteins tab. Clicking on an accession hyperlink
will load a Protein View report.
If the search included quantitation using Reporter
or Multiplex protocols, protein level quantitation information is available,
as in the following example, which comes from an iTRAQ experiment.
To select and re-order columns, expand the Columns section.
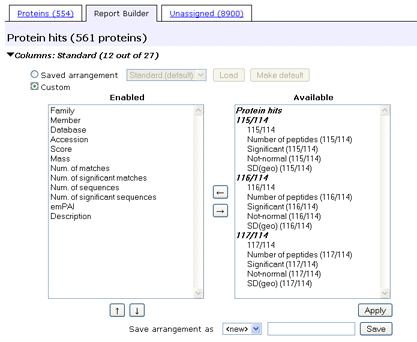
You can choose to load a saved arrangement or customise the table
by moving columns between the two
lists. The columns are categorised into groups. The basic set of columns that are always
available are under "Protein hits". Quantitation results create a set of columns
for each reported ratio.
In the enabled list, you can select individual columns, CTRL+click multiple
columns, or SHIFT+click a range
of columns and use the up and down arrows to change their relative position.
Here, we've added some quantitation columns and removed columns that were not
so relevant.
The changes take effect when you choose Apply.
If Mascot security is enabled and there is an arrangement you might want to use again,
you can save the arrangement in your security session.
Give it a name and choose Save. Saved arrangements can be loaded using
the drop down list towards the top.
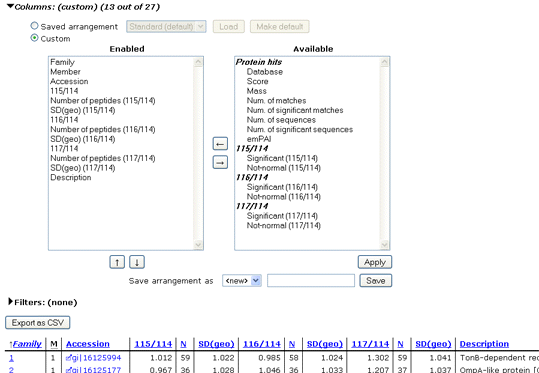
The Filters section enables protein rows to be dropped according
to multiple criteria. Configure the first term and apply it by choosing Filter.
The table will be reloaded and additional controls displayed to allow another
term to be added, if required. Often, a single term requiring each protein to have
significant matches to at least two distinct sequences will be all that is needed.
On the other hand, for a quantitation report, it is might be very useful to create a table
such as this, limited to proteins that are significantly up-regulated in the 117 channel
and significantly down-regulated in the 116 channel.
The unassigned list contains peptide matches that are not assigned to protein families.
In some cases, there may be no match at all, and only the observed m/z value and the experimental Mr will
be listed against the query number. In other cases, details of the top scoring peptide match will be listed.
The list is split into pages. Controls at the top and bottom of the tab can be used to
move between pages and change the number of matches per page. The Sort unassigned control allows the sort order to be changed.
Descending score makes it easy to see whether there are any good matches. If so,
you will want to increase the number of protein families or set it to AUTO so as to pull these matches
into the main body of the report. Ascending query number is the same as ascending precursor Mr.
Descending intensity allows you to find spectra with intense peaks that have failed to get a match. These could
be candidates for de novo sequencing.
The unassigned table contains the following columns:
- Query number, hyperlinked to Peptide View.
- Experimental m/z value
- Experimental m/z transformed to a relative molecular mass
- Relative molecular mass calculated from the matched peptide sequence
- Difference (error) between the experimental and calculated masses
- Number of missed cleavage sites
- Ions score
- Expectation value for the peptide match. (The number of times we would expect to obtain an equal or
higher score, purely by chance. The lower this value, the more
significant the result).
- Rank of the peptide match, (1 to 10, where 1 is the best match). This will always be 1 for
matches in the unassigned list. If there is a triangle
to the left of the rank, the row can be expanded to show the alternative
matches for this query
- Sequence of the peptide in 1-letter code with modified residues underlined.
- Any variable modifications used to obtain the match
When you load the peptide view for an unassigned query, it takes the first protein containing the matched
peptide. This may or may not be the protein that would be selected as the anchor protein if the formatting
was changed in a way that caused the match to be pulled into a family in the Proteins tab.
At the top of the tab, there are text search controls.
You can search the unassigned list by
query number, mass, m/z value, and peptide sequence. Select the category, enter text
in the edit field and choose Filter.
If the text is found, the complete unassigned
list is filtered to display matching queries. Choose Clear to return to the standard
display. If the text is not found, a button provides the option to try the search
in the Proteins tab.
Sections of the report are described in the order in which they appear.
Use
this link to open an example report in a new browser window or tab.
At the top of the report are a few lines to identify the search uniquely:
search title, date, user name, etc. The database version is identified with either a release
number or an ISO datestamp. The accessions and descriptions for the top scoring protein
hits are listed.
If the search included the auto-decoy option,
false discovery rate information is displayed at this location.
Following the header, a histogram illustrates the protein score distribution. The
50 highest protein scores are divided into 16 bins according to their score,
and the heights of the bars show the number of matches in each bin.
For a search of MS/MS data, protein scores are derived from ions scores as a
non-probabilistic basis for ranking protein hits. The protein score histogram has
little meaning for MS/MS results, but is retained for historical reasons. The green shaded area
extends up to the average identity threshold for an individual peptide match.
These controls enable the report format to be modified. After making changes, press the
"Format As" button to reload the report using the new settings.
- Report format Choose from the list of available formats
- Significance threshold The default significance threshold is p < 0.05. You can
change this to any value in the range 0.99 to 1E-18.
- Maximum number of hits This value was initially chosen when the search was submitted.
Enter a positive integer if you wish to re-specify the number of protein hits to report.
Of course, the total number of hits actually found by the search may be less.
Entering the word AUTO or a value of 0 will display all of the hits
that have a protein score exceeding the average identity threshold score for an individual peptide match.
- Standard or MudPIT scoring MudPIT scoring is a more aggressive protein score that removes
protein hits that have high protein scores purely because they have a large number of low-scoring
peptide matches. It is the default protein
score for a search in which the ratio of the number of queries to the number of entries in the database,
(after any taxonomy filter), exceeds 0.001.
- Ions score cut-off Values greater than 0 and less than 1 act as an expect value threshold,
and the scores for any peptide matches
with higher expect values are suppressed. Values of 1 or more act
as a score threshold, and any peptide matches with lower scores are suppressed. By setting this to (say) 20,
you cut out all of the very low scoring,
random peptide matches. This means that homologous proteins are more likely to collapse into a single hit.
- Show sub-sets By default, each hit in a Peptide Summary shows the set of proteins that match a
particular set of peptides. Proteins that match a sub-set of those peptides are not shown. You can choose
to show these additional protein hits, but be aware that this can make the report very much longer.
To show all sub-set proteins, set Show sub-sets to 1. To show no sub-set proteins, set it to 0.
Intermediate values set a threshold on the difference in protein score between the primary hit
and the sub-set hit expressed as a fraction. For example, if the protein score of the primary hit
was 400, and Show sub-sets was 0.75, sub-set hits with scores of 100 or more would be
displayed.
- Show or Suppress pop-ups The JavaScript pop-up windows, that show the top 10 peptide matches
for each query, are very useful, but they make the HTML report larger and slower to load in a web
browser. If you have a report that never seems to load, or is very slow to scroll, try suppressing the
pop-ups.
- Sort unassigned These are sorting options for the list of peptide matches that are not
assigned to protein hits. Descending score makes it easy to see whether there are any good matches. If so,
you will want to increase the number of protein hits or set it to AUTO so as to pull these matches
into the main body of the report. Ascending query number is the same as ascending precursor Mr.
Descending intensity allows you to find strong spectra that have failed to get a match. These could
be candidates for de novo sequencing.
- Require bold red Requiring a protein hit to include at least one bold red peptide match
is a good way to remove duplicate homologous proteins from a report. See the discussion
in Results Interpretation for further information.
The following controls are only displayed for certain types of search:
- UniGene index (Only displayed for a search of a nucleic acid database when UniGene index files have
been configured) Choose the UniGene index to be used to cluster the protein hits
into gene based families.
- Hide error tolerant matches (Only displayed for an automatic error tolerant search)
Check to hide the additional, error tolerant matches.
- Show Percolator scores (Only displayed for searches that satisfy the requirements for using
Percolator) Check to display scores and expect values adjusted by Percolator.
If the search used a quantitation method specifying Multiplex or Reporter protocol,
there will be an additional block of quantitation related controls, as
described here.
A search can easily be repeated, so as to investigate the effect of changes in search
parameters. Queries can selected in the result report, then loaded into a search form,
where the search parameters can be modified.
Checkboxes for selecting queries for a repeat search are included in the body of the report, wherever
the top rank match fir a particular query first appears. You can toggle individual checkboxes or use the
Select All and Select None buttons to change the states of all checkboxes. Search
Selected invokes the search form.
There is a second series of checkboxes, one for each protein hit, that have a dual purpose.
If you wish to perform a manual error tolerant search,
first check the Error Tolerant checkbox then select the proteins to be searched in the second pass
search. Press Search Selected to invoke the search form.
If the Error Tolerant checkbox is not checked, the checkboxes select protein hits for an
Archive Report
The body of the Peptide Summary report contains a tabular listing of the proteins, sorted by
descending protein score. For each protein, the first line contains the accession string,
(linked to the corresponding Protein View), the
protein molecular mass, and the protein
score. The number of queries matched to the protein
completes the first line. The second line is the protein description taken from the
Fasta entry. This is followed by a table
summarising the matched peptide masses. The table columns contain:
- Checkboxes
for selecting queries for a repeat search will appear in the first column of
any row containing the the first appearance of a top ranked match.
- Query number, hyperlinked to Peptide View.
- Experimental m/z value
- Experimental m/z transformed to a relative molecular mass
- Relative molecular mass calculated from the matched peptide sequence
- Difference (error) between the experimental and calculated masses
- Number of missed cleavage sites
- Ions score - If there are duplicate matches to the same peptide, then
the lower scoring matches are shown in brackets.
- Expectation value for the peptide match. (The number of times we would expect to obtain an equal or
higher score, purely by chance. The lower this value, the more
significant the result).
- Rank of the ions match, (1 to 10, where 1 is the best match).
- A letter U if the peptide sequence is unique to the protein hit
- Sequence of the peptide in 1-letter code. The residues that
bracket the peptide sequence in the protein are also shown, delimited
by periods. If the peptide forms the protein terminus, then a dash
is shown instead.
- Any variable modifications found in the peptide
If the search used a quantitation method specifying Multiplex or Reporter protocol,
there will be an additional rows and columns of quantitation information, as
described here.
An abbreviated listing follows for any proteins that contain the same set of peptide matches.
It is also possible to display proteins containing a sub-set of peptide matches, but this
is disabled by default. It can be enabled globally in the configuration file, mascot.dat, or
enabled for a single report by using the checkbox in the format controls.
Clicking on the query number link opens the Peptide View for the match
in a new browser window or tab. Resting the mouse cursor over the query number link causes a pop-up window
to appear, displaying the complete list of peptide matches for that query.
The pop-up window displays the query title (if any) followed by one or two
significance thresholds, which are described in detail
here. Below this, a table containing
information on the highest scoring peptide matches for the query:
- Ions score
- Expect value
- Difference (error) between the experimental and calculated masses
- Hit number of the (first) protein containing the peptide match. A plus sign
indicates that multiple proteins contain a match to this peptide
- Accession string of the (first) protein containing the peptide match.
- Sequence of the peptide in 1-letter code. If a variable modification has been used
to obtain a match, the modified residue is underlined.
If the residues that bracket the peptide sequence are the same in all the proteins that
contain it, then these residues are also shown, delimited
by periods. If the peptide forms the protein terminus, then a dash
is shown instead.
The unassigned list contains peptide matches that are not assigned to proteins in the body of the report.
In some cases, there may be no match at all, and only the observed m/z value and the experimental Mr will
be listed. In other cases, the top scoring peptide match will be listed.
So, unassigned doesn't necessarily mean unmatched or not significant. Its the overflow, if you like.
If you reformat the report, asking for more and more protein hits, all of the unassigned matches with
non-zero scores would eventually get pulled into the body of the report.
When you load the peptide view for an unassigned query, it takes the first protein containing the matched
peptide. This may or may not be the protein that would be selected as the primary hit if the peptide
match was pulled into the body of the report.
At the foot of the report, the search parameters are summarised. Descriptions
of individual search parameters can be found here.
Use
this link to open an example report in a new browser window or tab.
The Select Summary was inspired by David Tabb's
DTASelect. It is very
similar to the Peptide Summary, but more compact because
multiple matches to the same peptide sequence are collapsed into a single line. Also,
the list of peptide matches that are not assigned to any protein hit is split off into a separate
report.
The differences between the Select Summary and the Peptide Summary are as follows:
- The Search Parameters are moved up into the header
- There is no Score Distribution histogram
- There are no checkboxes against protein hits or peptide matches. The choices for
repeating a search are set by radio buttons for All queries, Unassigned, Below homology threshold,
and Below identity threshold. This means that you cannot use a Select Summary for
a manual error tolerant search or to create an
Archive Report
- In the Protein Hit List, if multiple queries match to the same peptide
(the same sequence, modifications and charge state) full details are displayed for the highest scoring
match only. Any matches with lower scores are listed as additional query number links after the peptide
sequence.
- Variable modifications are not listed after the peptide sequence, but are indicated by
underlined residues.
- Unassigned Peptide Matches are split off into a separate report.
Use the format controls to display this report.
Use
this link to open an example report in a new browser window or tab.
An Archive Report is a Peptide Summary in which only selected protein hits appear.
There is no unassigned list and no controls for changing the format or repeating the search.
You might use this format to give a colleague a list of validated protein hits, or to remove hits
that are contaminants or of no interest, etc.
|