Decoy Databases
Many journals impose guidelines for the reporting of database search results, designed
to ensure that the data are reliable.
This was initiated by the Editors of Molecular and Cellular Proteomics, who organised
a workshop in 2005 to discuss the issues, culminating in the
"Paris Guidelines". The current
guidelines require "For large scale experiments, the
results of any additional statistical analyses that estimate a measure of identification
certainty for the dataset, or allow a determination of the false discovery rate, e.g.,
the results of decoy searches or other computational approaches."
This is a recommendation to repeat the search, using identical search parameters, against a
database in which the sequences have been reversed or randomised. You do not expect to get any
true matches from the "decoy" database. So, the number of matches that are found is an
excellent estimate of the number of false positives that are present in the results from the real
or "target" database. This approach has been described in publications from Steven
Gygi's group, e.g. Elias, J. E., et al., Comparative evaluation of mass spectrometry platforms used
in large-scale proteomics investigations, Nature Methods 2 667-675 (2005).
If TP is true positive matches and FP is false positive matches, the number of matches in
the target database is TP + FP and the number of matches in the decoy database is FP.
The quantity that is reported is the False Discovery Rate (FDR) = FP / (FP + TP)
While this is an excellent validation method for MS/MS searches of large data sets, it is not
useful for a search of a small number of spectra, because the number of matches is too small
to give an accurate estimate. Hence, this is not a substitute for a reliable scoring scheme,
it is more a good way of calibrating it.
A decoy search can be performed automatically by choosing the
Decoy checkbox on the search form. If you prefer to create
a decoy database and search it separately, a utility for this purpose is available
below.
For an automatic decoy database search, choose the Decoy checkbox on the search form.
During the search, every time a protein
sequence from the target database is tested, a decoy sequence of the same
length is automatically generated and tested. The average amino acid composition of the decoy
sequences is the same as the average composition of the target database.
The matches and scores for the decoy sequences are recorded separately
in the result file. When the search is complete, the numbers of matches and
the false discovery rate are reported in the result header.
This screenshot shows an example of the decoy statistics for an MS/MS search as displayed
in the Protein Family Summary. The same information is displayed
with slightly different formatting in the Select and Peptide Summaries:

In most cases, the Mascot identity threshold is over-conservative, and better sensitivity
at a given false discovery rate will be achieved by using the homology threshold.
In this example, the significance threshold is the default, 0.05, yet the false discovery rate
for matches above the homology threshold is below 5%. The buttons and drop down
lists to the right (Protein Family Summary only) can be used to
automatically adjust the significance threshold so as to achieve a specified
false discovery rate. If we choose 5% for the homology threshold and Filter,
the significance threshold is shifted to 0.071 to give the best possible sensitivity
at a false discovery rate less than or equal to the requested value.

The choice of values in the drop down list is a global setting in the options section of
mascot.dat.
TargetFDRPerCent 0.1, 0.2, 0.5, 1+, 2, 5
The numbers are percentages and the plus sign doesn't appear in the drop down list, it indicates the default
selection. Clicking on the Decoy link will load a report for the decoy search, just as if it was a
separate search of a decoy database.
Decoy statistics are always calculated from all matches. Changing the number of hits to be displayed
or setting a cut-off on the ions score or expect value will have no effect. Sometimes, it will not be
possible to achieve the requested false discovery rate; the significance threshold will be pushed
to its limit and a warning displayed. The automatic decoy search is equivalent to searching
separate target and decoy databases.
Most of the algorithms for constructing decoy sequences described in G. Wang, et al. (2009), "Decoy Methods for Assessing False Positives
and False Discovery Rates in Shotgun Proteomics", Anal Chem. 81(1):146-159
have been implemented. The two most popular are:
- Method 1: reversed protein sequences
- The default for MS/MS searches with a fully specific or semi-specific enzymes
- Method 3: randomised protein sequences
- The default for MS/MS searches with enzyme None and all PMF searches.
This was the default method in Mascot 2.3 and earlier. If the target database is nucleic acid, it is the
original sequence that is randomised, not the translation.
The defaults are specified in the options section of mascot.dat. Refer to the Setup & Installation
Manual for further details.
Conventionally, a decoy database search is only used for validating searches of MS/MS data.
It is not possible to get a false discovery rate for a peptide mass fingerprint, but it can be
informative to see the result of repeating a PMF search against a decoy database, especially
if the match from the target database is close to the significance threshold, or if there
is reason to think the experimental values or search parameters may be producing a false positive.
This screenshot shows an example of the decoy report for a PMF search:

A Perl script to reverse or randomise database entries can be downloaded here:
decoy.pl.gz. Unpack using
gzip or WinZip.
Note: Windows file associations can cause this file to be unpacked automatically
when downloaded using Microsoft Internet Explorer on a Windows PC. If you cannot
open the file in Winzip, try to open it in a text editor like WordPad. If it looks
like text, then it has been unpacked, and you only need to rename the file to decoy.pl.
Execute
without arguments to get the following instructions.
Usage: decoy.pl [--random] [--append] [--keep_accessions] input.fasta [output.fasta]
- If --random is specified, the output entries will be random sequences
with the same average amino acid composition as the input database.
Otherwise, the output entries will be created by reversing the input
sequences, (faster, but not suitable for PMF or no-enzyme searches).
- If --append is specified, the new entries will be appended to the input
database. Otherwise, a separate decoy database file will be created.
- If --keep_accessions is specified, the original accession strings will
be retained. This is necessary if you want to use taxonomy and the
taxonomy is created using the accessions, (e.g. NCBI gi2taxid).
Otherwise, the string ###REV### or ###RND### is prefixed to each
original accession string.
- You cannot specify both --append and --keep_accessions.
- An output path must be supplied unless --append is specified.
- If the database is nucleic acid, no need to specify --random. A
simple reversal will effectively randomise the translated proteins
Title line processing assumes that the accession string is between the ">" character
and the first white space. If this is not the case, you may need to edit the script to make it usable.
If creating a concatenated database, the Mascot parse rules will probably need to be
rules 4 and 5 if they are to work for both original and decoy entries. This makes it difficult to
configure taxonomy.
The Mascot report scripts cannot display the match counts and FDR after a manual decoy search.
One option is to export the results to Excel using the custom CSV format. To avoid outputting duplicate matches when a query matches more
than one protein, make sure to set the number of hits to 1, include the unassigned list, and delete any
matches with rank greater than 1.
If using a concatenated database,
an easier alternative is to use a simple Perl script that can be downloaded here:
fdr_stats.pl.gz. Unpack using
gzip or WinZip.
Note: Windows file associations can cause this file to be unpacked automatically
when downloaded using Microsoft Internet Explorer on a Windows PC. If you cannot
open the file in Winzip, try to open it in a text editor like WordPad. If it looks
like text, then it has been unpacked, and you only need to rename the file to fdr_stats.pl.
Copy the script to the Mascot bin directory and execute without arguments to get the following usage instructions:
Output counts of matches for a specified FDR
The program must be run from the mascot bin directory
Usage: fdr_stats.pl fdr_goal thresh_type decoy_string result_file [debug]
Example: fdr_stats.pl 0.01 homology "DECOY_" ../data/20111213/F123456.dat
fdr_goal is the desired peptide FDR (enter 0.01 for 1%)
thresh_type is either identity or homology
decoy_string is the substring in a protein accession that identifies a decoy entry
result_file is the path to a Mascot result file
add optional final argument "debug" to get details of all matches (tab separated)
The Gygi group advocate searching a database in which the target and decoy sequences have been
concatenated. This means that you will only record a false positive when a match from the decoy
sequences is better than any match from the target sequences. A more conservative approach is to
search the two databases independently. If the Mascot score threshold for a given spectrum is
(say) 40, and we get a match of 60 from the target database and 50 from the decoy database, this
would not count as a false positive from a concatenated database, but it would count as a false
positive if the two had been searched independently.
There is also the question of whether to reverse or randomise. If you simply reverse a sequence,
and then do the search without enzyme specificity, you may get a misleading picture of the false
positive rate because, sometimes, you will get a mass shift at each end of a reversed peptide that
just happens to transform a genuine y series match into a false b series match or vice versa.
Similarly, a reversed database is not suitable for verifying a peptide mass fingerprint score,
because half of the tryptic peptide mass values will be unchanged. (Those that have the same
residue at the C-terminus and flanking the N-terminus). The main objection to using a randomised database is that the number of distinct peptide sequences
in the decoy is likely to be larger than in the target because real protein sequences
have a degree of redundancy, which is lost on randomisation.
The performance of a scoring scheme is sometimes illustrated as a Receiver-Operating
Characteristic or "ROC Curve". This plots true positive rate and false positive
rate as a function of a discriminator, such as a score threshold.
A good scoring scheme will try to follow the axes, as illustrated by the red curve, pushing
its way up into the top left corner. A useless scoring algorithm, that cannot distinguish
correct and incorrect matches, would follow the yellow dashed diagonal line.

The origin of the ROC
curve has unit specificity, i.e. zero false positives, but also zero true positives.
Not a useful place to be. The top right of the ROC curve has unit sensitivity, i.e. 100%
true positives, but also 100% false positives, which is equally useless. By setting a
significance threshold in Mascot, you effectively choose where you want to be on the curve.
A ROC curve is designed to illustrate a so-called binary classifier. In our case, an
MS/MS spectrum either represents a peptide in the database or it does not. The search engine
is the classifier, which either succeeds or fails to report the correct match.

To plot an authentic ROC curve, we need estimates of the numbers of true negatives (TN) and
false negatives (FN), because true positive rate = TP / (TP + FN) and false positive rate =
FP / (FP + TN). However, for real-life datasets, where we are dealing with unknown samples,
we do not know TN and FN. So, what is presented as a ROC curve is usually just a plot of the
fraction of spectra matched in the target database versus the fraction matched in the decoy,
or something similar.
In most searches, a proportion of the spectra are
unmatchable for all sorts of reasons. Some spectra are non-peptidic, or little more than noise; others cannot be matched
because the sequence is not in the database or the peptide is modified in a way that is not part
of the search. If you plot a ROC-style curve for the typical MudPIT data set, where it is quite normal for
90% or more of the spectra to be unmatchable, you will get a very poor looking curve,
because no scoring scheme can discriminate the unmatchable spectra. In other words, as the score threshold is
reduced towards zero, additional matches are equally likely to come from the decoy as from the target, and the
ROC curve tends towards a diagonal line, as shown in the first plot.

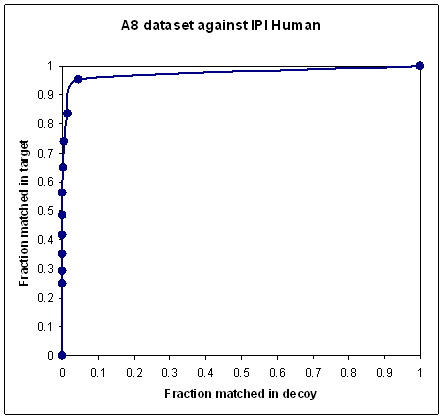
To obtain a nice looking curve, like the second plot, you must somehow exclude the unmatchable spectra.
This is where the problem lies. Deciding which spectra to exclude can be somewhat arbitrary. Clearly, if you reduce
a dataset to a handful of the highest quality spectra, then any scoring scheme will give a beautiful curve.
So, a nice looking curve by itself doesn't prove that a scoring scheme is any good;
it may just be the result of cherry-picking the higher quality spectra.
|